Getting Started
NOTE: Both OpenCravat and OakVar can be used to annotate a human genome. At the beginning of the project we used OpenCravat as a framework. However, as OakVar is based on OpenCravat and contains more advanced features customized specially for personal longevity genomics, we decided to base further development of the project on OakVar.
Installing OakVar
Since our module is based on OakVar, you have to install OakVar first to run our module, if it is not already installed. OakVar docs: https://oakvar.readthedocs.io/en/latest/
- Pre-requirements for Oakvar:
installed conda/mamba environment management systems, or you can use their lighter versions: miniconda/micromamba
installed python and pip
You can find documentation for mamba here: https://mamba.readthedocs.io/en/latest/index.html
And for conda here: https://docs.conda.io/en/latest/
The installation of OakVar and the further work should proceed after activation of an environment created by Conda/Mamba or Miniconda/Micromamba.
Installing Annotators
For the Longevity module to work, you need to install the following annotators:
clinvar
dbsnp
gnomad
ncbigene
omim
pubmed
longevitymap
You can install them by using terminal or Oakvar GUI.
Installation using terminal:
Use the following command:
ov module install module_name
Installation using GUI:
To activate Oakvar GUI, use the following command:
ov gui
After the execution GUI will be opened in your browser.
Go to “Store”:
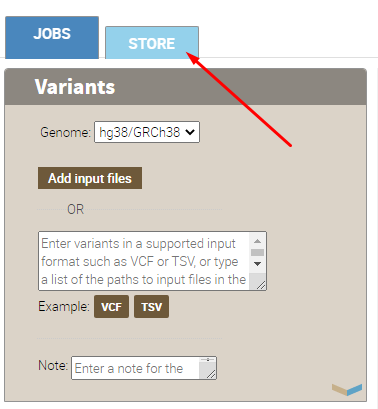
Find annotators and install them:
Installing the Reporter
Installation using terminal:
Use the following command in terminal:
ov module install longevity-combinedreporter
Installation using GUI:
To activate Oakvar GUI, use the following command:
ov gui
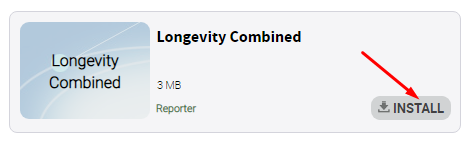
Go to “Store”:
Find the reporter called “longevity-combinedreporter” and install it:
Loading Genome Files
Open OakVar in your browser. You will see the index page:
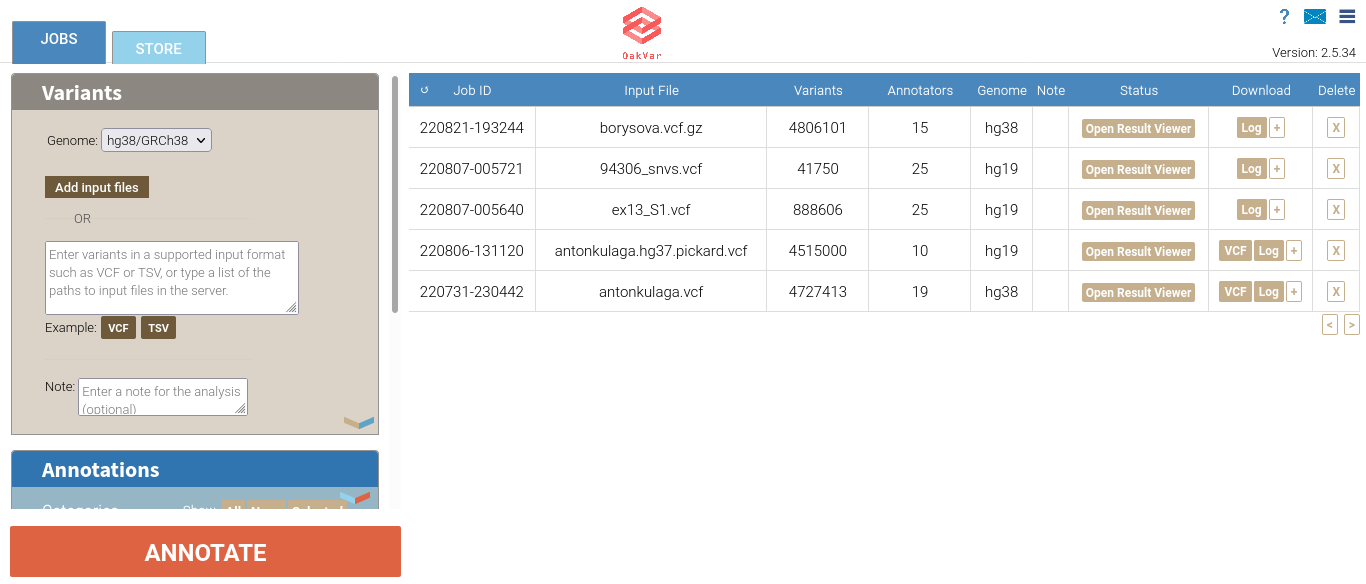
In the Variants section you should choose the right assembly version of the Genome: hg38/GRCh38, hg19/GRCh37, or hg18/GRCh36.
For example we’ll take a small VCF file of the hg19/GRCh37 version named example.vcf.
Click Add input files. A file upload dialog will open, allowing to browse and select the vcf file (or multiple files at once).
After loading, the file(s) will be shown next to the Add input files button along with another button Clear file(s) and a small X button next to each file name. If you click that X, the appropriate file will be deleted. If you click Clear file(s), all the files you loaded will be deleted.
Installing and Selecting Necessary Annotators
Scroll the left area down to the Annotations section.
Here you can see the categories of annotators available for selection (above) and checkboxes for particular annotators.
Annotators are software modules which can be developed, added and installed as needed. If any necessary annotator is not yet installed, you can install it on the STORE tab in the upper left corner.
All annotators can be divided into 2 groups:
Tools that predict pathogenicity (bold)
Tools that provide information like databases
Here are their internal (coded) module names:
cadd_exome (1.6.1) - CADD is a tool for scoring the deleteriousness of single nucleotide variants as well as insertion/deletions variants in the human genome
gnomad_gene (2.2.1) - gene level population statistics from gnomAD
pubmed (1.1.5) - articles related to a particular gene
clingen (1.0.1) - NIH-funded resource that defines the clinical relevance of genes and variants
clinpred (1.0.0) - prediction tool to identify disease-relevant nonsynonymous single nucleotide variants
clinvar (2021.10.01) - ClinVar is an archive of reports of the relationships among human variations and phenotypes, as well as interpretations of clinically relevant variants (Uncertain significance, Likely pathogenic, Pathogenic etc.)
mitomap (1.1.0) - a human mitochondrial genome database
ncbigene (2019.08.02) - gene descriptions from NCBI (National Center for Biotechnology Information) Gene database
omim (1.0.0) - catalog of human genes and genetic disorders and traits
prec (3.6.0) - provides a database identifying rare and likely deleterious loss-of-function (LoF) alleles
provean (1.0.0) - tool which predicts whether an amino acid substitution or indel has an impact on the biological function of a protein
revel (2020.12.02) - ensemble method for predicting the pathogenicity of missense variants based on a combination of scores from 13 individual tools
sift (1.2.0) - predicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids
GnomADD - aggregating and harmonizing both exome and genome sequencing data from a wide variety of large-scale sequencing projects
PharmGKB - an NIH-funded resource that provides information about how human genetic variation affects response to medications
dbSNP - the Single Nucleotide Polymorphism Database is a free public archive for genetic variation within and across different species developed and hosted by the National Center for Biotechnology Information (NCBI) in collaboration with the National Human Genome Research Institute (NHGRI)
Once an annotator is installed, you can select in on the JOBS tab in the upper left corner.
For example, let’s select the ClinVar annotator from the Clinical Relevance category:
Note: An annotator may belong to multiple categories at once.
The checkbox and X buttons between the categories and the annotators sections allow to select all of the displayed annotator chechboxes or to clear all of them.
If you right-click any annotator, a pop-up window with its description will open in the right area:

For our purposes we will need the following annotators: ClinVar (clinvar), dbSNP (dbsnp), gnomAD3 (gnomad), LongevityMap (longevitymap), NCBI Gene (ncbigene), OMIM (omim), and PubMed (pubmed). If any of them are missing, install them on the STORE tab, then go back to JOBS, in the Annotations section select All categories, and then select each of the annotator checkboxes.
Annotating Your Genome
When you select all the annotators you need, click the large ANNOTATE button below in the left area.
Annotating a large genome file may take some time. While loading, it will appear in the right area on the top of the list, displaying different stages of the processing in the Status column, and when finished, the Open Results Viewer button will appear in that column of the particular genome row:
Opening Your Annotated Genome
Now click the Open Results Viewer button, and the annotated genome will open in a new browser tab/window.